![]()

For information on using the package, see the stable documentation. Use the in-development documentation for the version of the documentation, which contains the unreleased features.
using NBodySimulator
using StaticArrays
using Plots
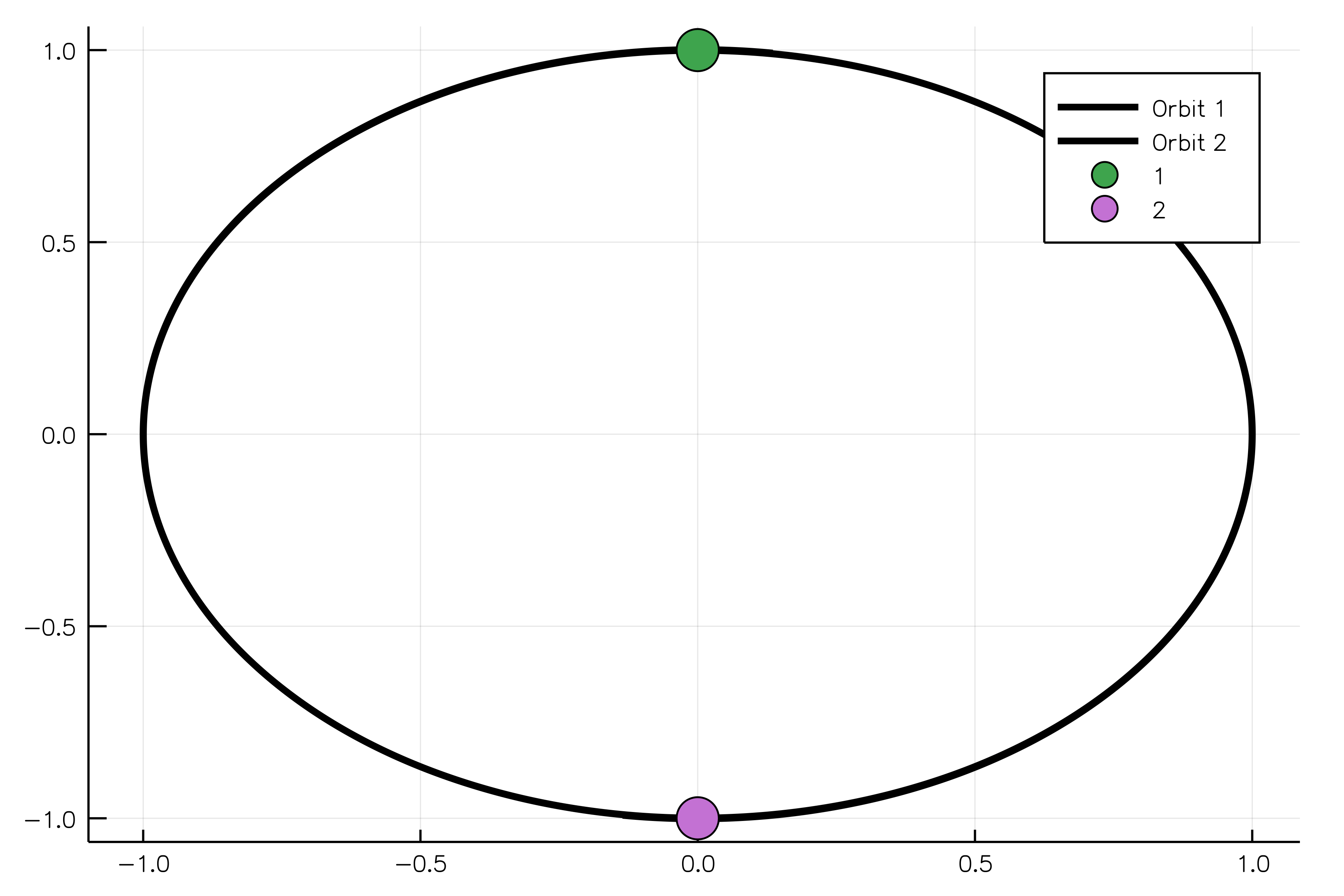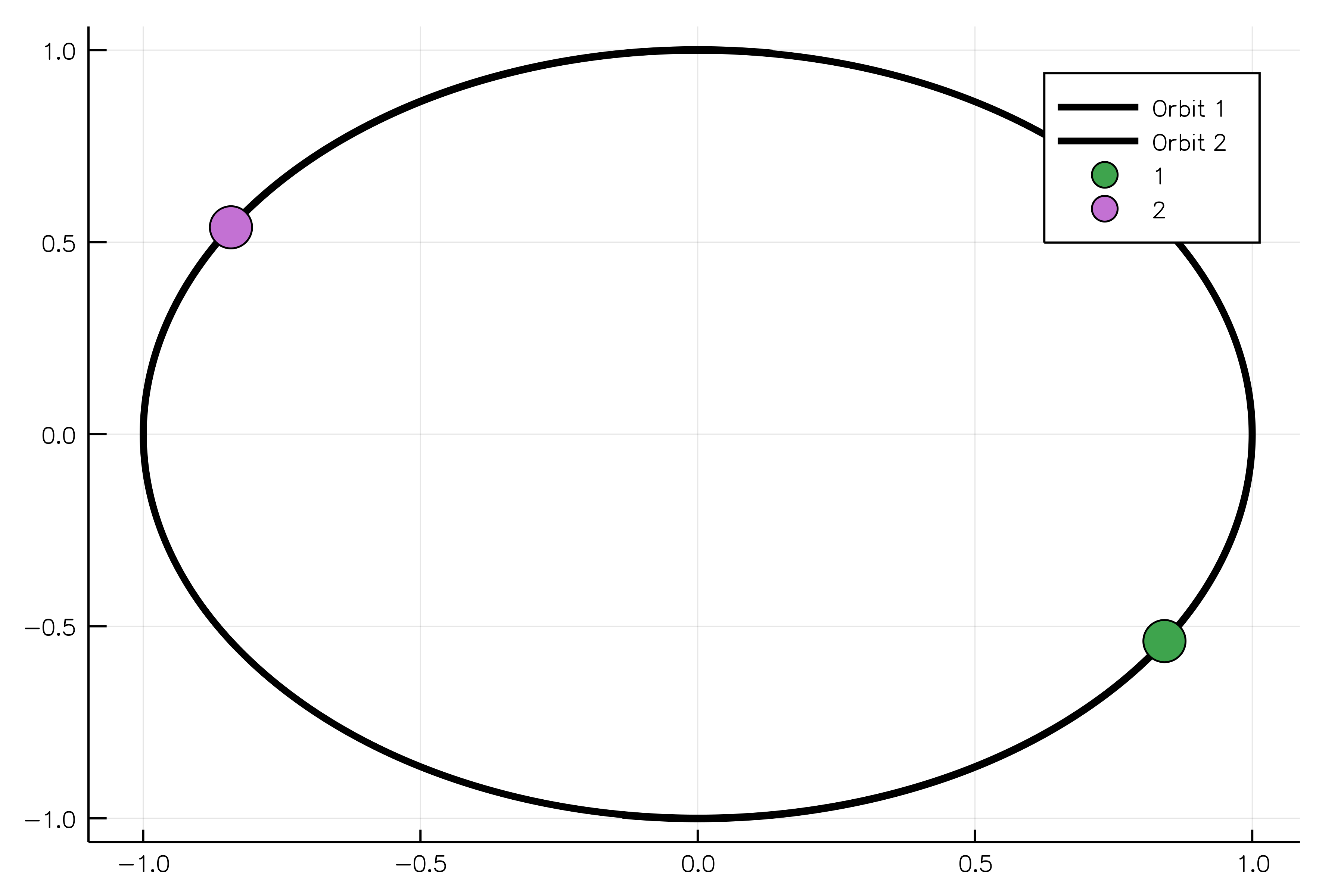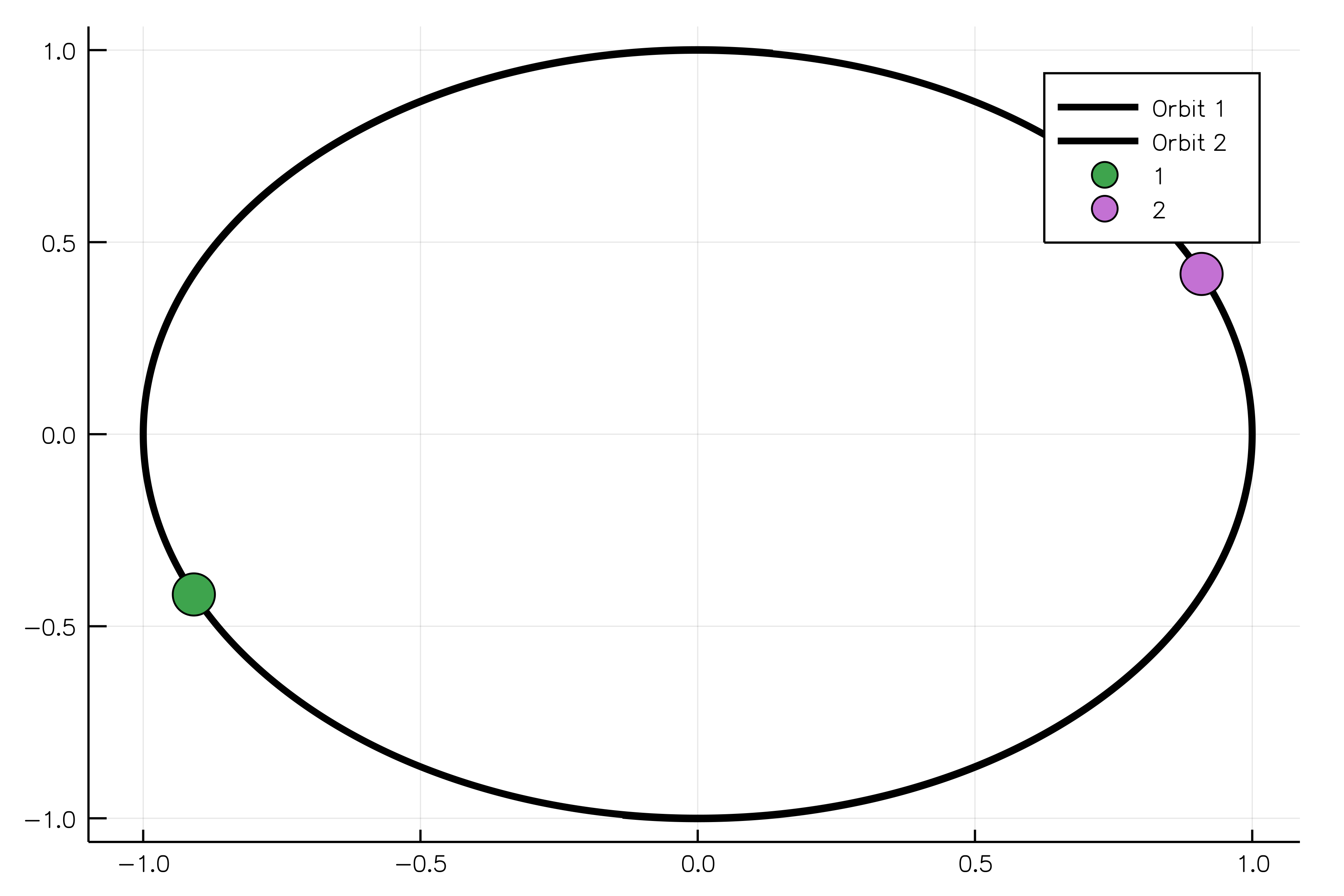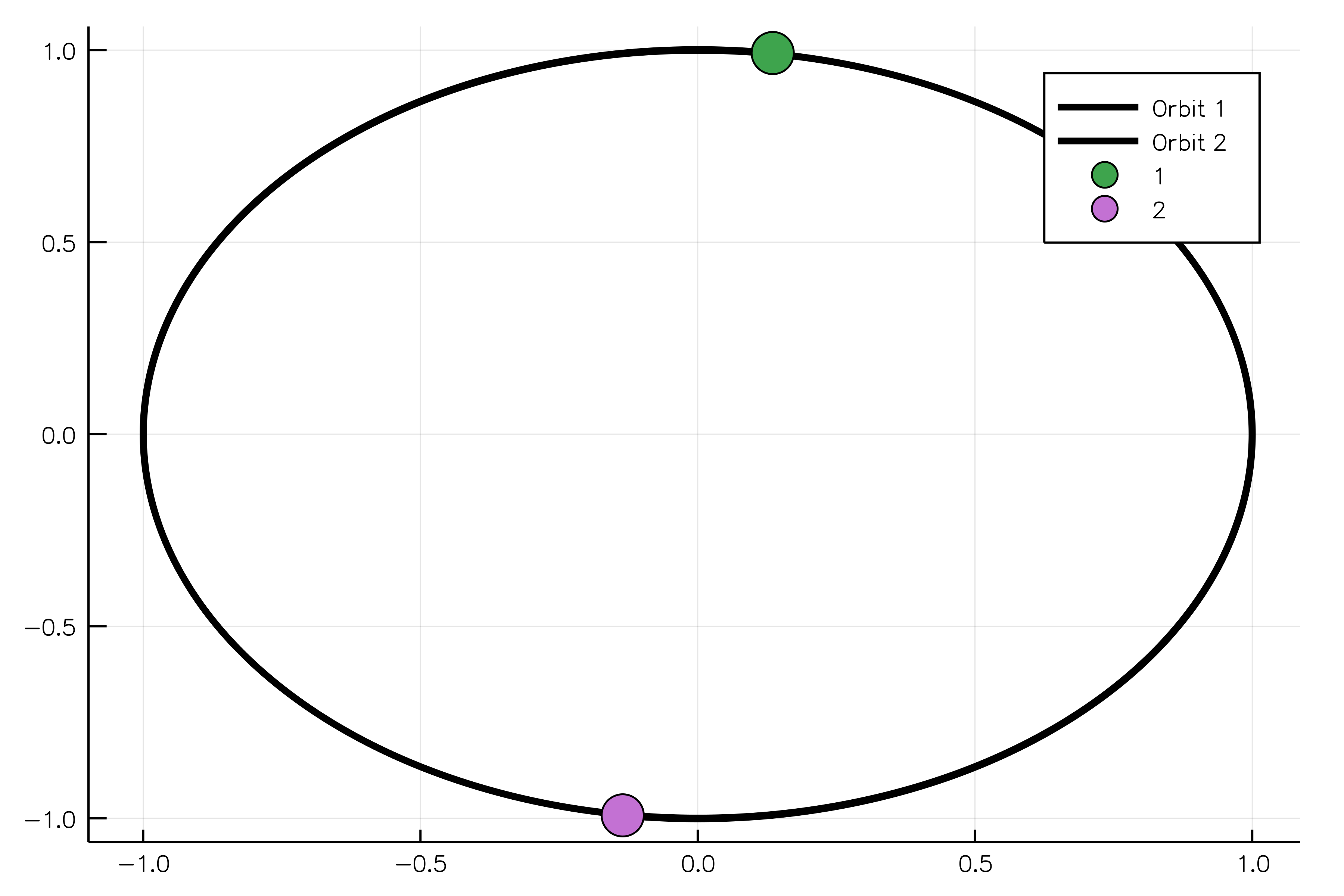
body1 = MassBody(SVector(0.0, 1.0, 0.0), SVector(5.775e-6, 0.0, 0.0), 2.0)
body2 = MassBody(SVector(0.0, -1.0, 0.0), SVector(-5.775e-6, 0.0, 0.0), 2.0)
G = 6.673e-11
system = GravitationalSystem([body1, body2], G)
tspan = (0.0, 1111150.0)
simulation = NBodySimulation(system, tspan)
sim_result = run_simulation(simulation)
animate(sim_result, "path_to_animated_particles.gif")